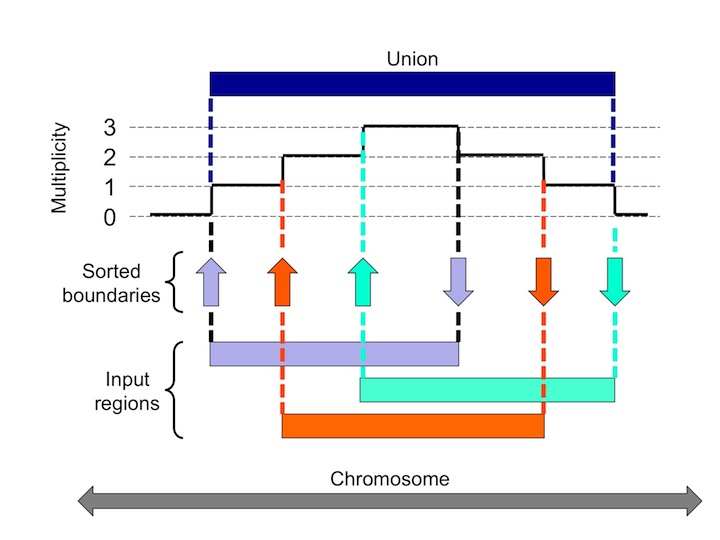
The MULTOVL suite of programs finds multiple overlaps among genomic regions. The basic idea is that the start and end coordinates of the regions on the same chromosome are sorted in increasing position and then we walk along this list and increase or decrease a counter whenever a start or end position is encountered, respectively.
The actual value of this counter indicates the multiplicity of the underlying overlap. The multiplicity is 0 outside the regions, 1 over those parts that do not overlap, 2 over pairwise overlaps, 3 over triples, ... etc. The sorting can be done in O(N log N) time, and the walking-over is linear, which makes the whole process very efficient.
The MULTOVL algorithm can detect several kinds of region overlaps. In addition to the N-fold overlaps you can detect solitary regions which are the input regions that do not overlap with any other region in the input data set, and union overlaps that is the union of the input regions that overlap at least once somewhere.
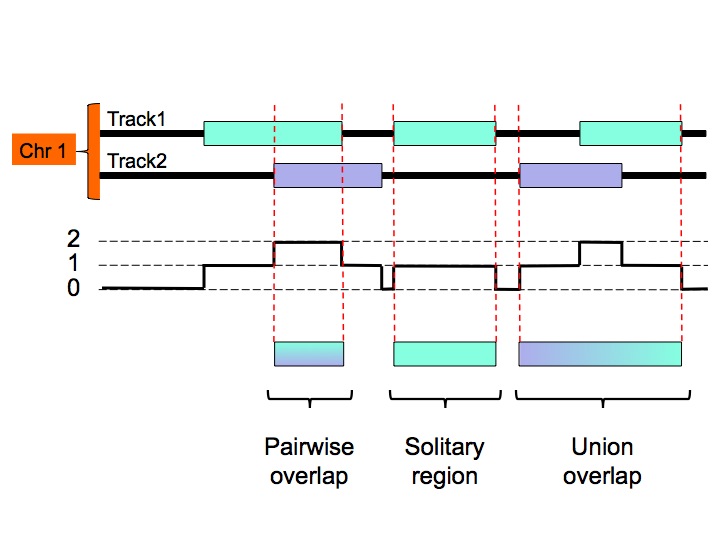
Which kind of overlaps are detected will be specified through command-line options.
Because overlaps make sense only between regions on the same chromosome, the MULTOVL algorithm can easily be parallelized as the overlap detection on a chromosome is fully independent from what's happening on another chromosome. Reading lots of huge input files can also be done in parallel in theory, leading to the following high-level schema:-
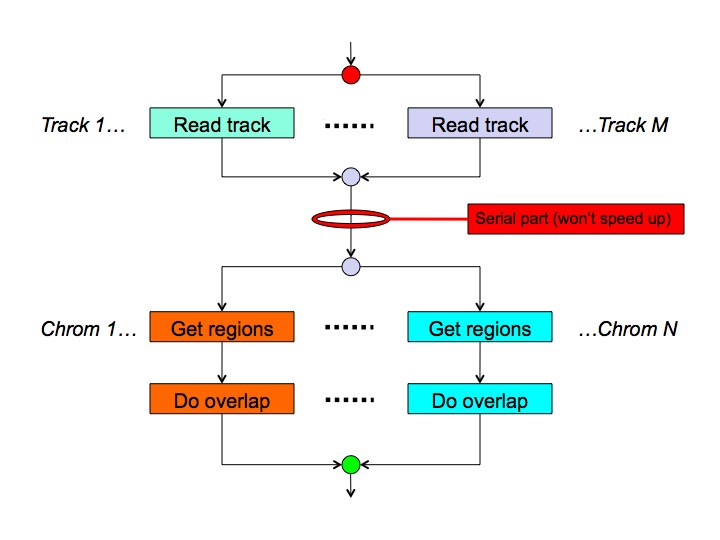
This is a planned feature which has not been implemented yet.
The MULTOVL algorithm and implementation have been published in the following Application Note:
Aszódi, A. (2012): MULTOVL: Fast multiple overlaps of genomic regions. Bioinformatics 28: 3318-3319.
Please include the above reference in scientific publications if you use MULTOVL.
Any decent C++ compiler supporting the C++98 version of the language can in principle be used, implementation quirks and potential incompatibilities notwithstanding. The following compilers have been used in building MULTOVL:
MULTOVL needs the Boost C++ libraries, Version 1.44 or above.
MULTOVL builds are configured with CMake, Version 2.8.8 or above. Documentation is generated with Doxygen, Version 1.7 or above.
Change to the top-level directory which will be called ${MULTOVL} from now on, make a directory called build, change to it and configure your build with CMake:
cd ${MULTOVL}
mkdir build
cd build
cmake -DCMAKE_BUILD_TYPE=Release \
-DCMAKE_C_COMPILER=your_favourite_C_compiler \
-DCMAKE_CXX_COMPILER=your_favourite_C++_compiler \
-DBOOST_ROOT=/path/to/boost \
-DCMAKE_INSTALL_PREFIX=/path/to/installdir ..
If the -DCMAKE_C_COMPILER or -DCMAKE_CXX_COMPILER parameters are omitted, the system default C and C++ compilers will be used.
CMake's Boost detection script is not perfect. Sometimes it is not enough to specify -DBOOST_ROOT: for instance on 64-bit OpenSuSE, the Boost headers are in /usr/include/boost but the libraries are in /usr/lib64. In this case you have to specify -DBOOST_INCLUDEDIR=/usr/include and -DBOOST_LIBRARYDIR=/usr/lib64 separately.
The libraries will be linked statically by default. You can ask CMake to link all external libraries dynamically by specifying the option -DMULTOVL_USE_STATIC_LIBS=OFF. Or you can specify for each external library whether you want to have static or dynamic linking by setting the options -D<lib>_USE_STATIC_LIBS=ON where <lib> can be Boost, BAMTOOLS.
Assuming the configuration went well, you can compile and install the package and its documentation:-
make all
make install
The tools will be installed in /path/to/installdir/multovl-1.3/bin.
Alternatively you can configure a debug build by specifying -DCMAKE_BUILD_TYPE=Debug and omitting the -DCMAKE_INSTALL_PREFIX setting when invoking cmake. This is important only for developers.
You can check the configuration and build details of the package by invoking the program multovl_config.
These instructions have been tested on Windows 10 using Visual Studio 2015. It is quite likely that they work under Windows 8.x or Windows 7, and possibly with Visual Studio 2013 as well. It is recommended to install a pre-compiled binary distribution of the Boost libraries.
The BamTools library, now integrated in Multovl, relies on the Zlib compression library which is installed on most UNIX systems by default but not on Windows. Building it in all necessary variants is very tedious, so I decided to include the important bits from the latest version (1.2.8) in the Multovl source as well. If CMake does not find a system Zlib then it will build the Multovl-supplied version.
Once you have CMake and Boost installed, launch a Visual Studio command prompt, navigate to the Multovl directory, create a build directory under it and run CMake:
mkdir build
cd build
cmake -G "Visual Studio 14" -DBOOST_ROOT=path\to\boost
-DBoost_LIBRARY_DIR=path\to\boost\lib32-msvc-14.0 ..
Open the Visual Studio solution multovl.sln and build Multovl as usual. Choose the debug or release versions from the Visual Studio toolbar as needed.
If you wish to build the 64-bit version then you have to invoke CMake a bit differently, instructing it to choose the 64-bit compilers amd the 64-bit Boost libraries:
mkdir build
cd build
cmake -G "Visual Studio 14 2015 Win64" -DBOOST_ROOT=path\to\boost
-DBoost_LIBRARY_DIR=path\to\boost\lib64-msvc-14.0 ..
The MULTOVL tools obey a few conventions. When invoked without arguments or with the -h or --help command-line switch, they print a short help and then exit.
The overlap multiplicity is controlled by the -c,-m,-M options. -c N requests exactly N-fold overlaps, the default being N=2 for pairwise overlaps. If you want to detect a range of multiplicities, say you have 7 input files and you want three-, four- and five-fold overlaps, then specify -m 3 -M 5 on the commandline. The special setting -M 0 means "any multiplicity". Union overlaps can be requested by specifying the -u switch.
-c 1 requests solitary region detection. Note that "solitaryness" is always relative to the set of input tracks: what you get as "solitary" regions is a list of regions that do not overlap with any other region within the current input dataset. Solitary region detection can also be requested by specifying -m 1. Depending on what you set for -M x, this will produce solitaries AND up to x-fold overlaps.
The minimal length of overlaps (in basepair units) that are detected as such can be set with the -L n option, with n=1 by default. Sometimes you want to get "really solid overlaps" by accepting only longer overlaps, e.g. by setting -L 50. This won't list overlaps that are shorter than 50 basepairs.
Input tracks may contain overlapping regions, and these will be detected by default. The -n (no intra-overlaps) switch reduces the clutter somewhat by filtering out those overlaps that occur only between regions on the same track. Complex overlaps within and between tracks will still be reported, with a multiplicity corresponding to the number of tracks participating in the overlap, rather than the number of ancestor regions (which will be larger). In such cases it is advisable to after-process the MULTOVL output according to your needs. Note that when solitary region detection (-c 1) or unions of overlapping regions (-u) are requested, then the -n switch is ignored.
You can request timing information by specifying the -t switch. This is interesting only for serial/parallel runtime comparisons, normal users should not use this (all the more so because no output will be generated).
Multiple Chromosome / Multiple Region Overlaps
Usage: multovl [options] [<infile1> [ <infile2> ... ]]
Accepted input file formats: BED, GFF/GTF, BAM (detected from extension)
<infileX> arguments are ignored if --load is set
Output goes to stdout, select format with the -f option
Options:
-h [ --help ] Print this help and exit
--version Print version information and exit
-c [ --common-mult ] arg Find exactly arg-fold overlaps (default 2), -c 1
detects 'solitary regions', overrides -m, -M
-L [ --ovlen ] arg Minimal overlap length (default 1), -c 1 forces -L 1
-m [ --minmult ] arg Minimal multiplicity of the overlaps (default 2), 1
detects solitary regions,too
-M [ --maxmult ] arg Maximal multiplicity of the overlaps, 0 (the
default) means no upper limit
-u [ --uniregion ] List union of overlapping regions instead of each
overlap, ignored if -c 1 is set
-n [ --nointrack ] Do not detect overlaps within the same track,
ignored if -c 1 or -u is set
-t [ --timing ] List execution times only, no region output
-s [ --source ] arg Source field in GFF output
-f [ --outformat ] arg Output format {BED,GFF}, case-insensitive,
default GFF
--save arg Save program data to archive file, default: do not save
--load arg Load program data from archive file, default: do not load
You should supply at least one input file in BED or GFF format unless --load is specified (see below). The output will be written to the standard output in GFF2 or BED format with ancestry annotations (it is a good idea to pipe it to a file). The second column ("source") will be set to "multovl" by default but you can change this using the -s sourcestr option.
It is possible to save the internal data structures of MULTOVL to a binary archive file using the --save option. Note that this does not save the results themselves or any other command-line option settings. The saved status can be read from the archive by another invocation of MULTOVL with the --load option. The idea is to save time on input parsing and internal data setup when examining multiple overlaps of the same set of tracks using different parameters.
The --save and --load options write and read binary archives using the Boost::Serialization library. These are optimized for speed and are not portable between different architectures.
Sometimes you want to know whether the overlaps you detected are "significant". The multovlprob program helps here by offering the option of randomly reshuffling some of the tracks several times to approximate a null distribution of the overlap lengths. By comparing the actual overlap lengths to the null distribution, one can get a feeling of whether the actual overlaps could have been "caused by chance" or the overlaps occurred much more often (or much less often) than what could be expected.
Region reshuffling is performed within "free regions". These represent genomic segments in which it is possible to detect enrichments at all (e.g. regions tiled in microarrays or regions visible to high-throughput sequencing). All input regions must fit within a free region, multovlprob checks this before the analysis. Regions that do not fit will be skipped and a warning is printed to standard error.
multovlprob distinguishes between "fixed" and "shufflable" tracks. You can fix tracks where shuffling makes no sense, e.g. annotation tracks containing genes or transcripts. By default all input tracks are "shufflable".
Overlap length statistics is generated for each possible multiplicity. It can happen that a given multiplicity was not present among the actual overlaps but detected during reshuffling.
These are the options of multovlprob:
Multiple Region Overlap Probabilities
Usage: multovlprob [options] file1 [file2...]
file1, file2, ... will be reshuffled, there must be at least one
Accepted input file formats: BED, GFF/GTF, BAM (detected from extension)
Output goes to stdout
Options:
-h [ --help ] Print this help and exit
--version Print version information and exit
-c [ --common-mult ] arg Find exactly arg-fold overlaps (default 2), -c 1
detects 'solitary regions', overrides -m, -M
-L [ --ovlen ] arg Minimal overlap length (default 1), -c 1 forces -L
1
-m [ --minmult ] arg Minimal multiplicity of the overlaps (default 2), 1
detects solitary regions,too
-M [ --maxmult ] arg Maximal multiplicity of the overlaps, 0 (the
default) means no upper limit
-u [ --uniregion ] List union of overlapping regions instead of each
overlap, ignored if -c 1 is set
-n [ --nointrack ] Do not detect overlaps within the same track,
ignored if -c 1 or -u is set
-t [ --timing ] List execution times only, no region output
-F [ --free ] arg Free regions (mandatory)
-f [ --fixed ] arg Filenames of fixed tracks
-r [ --reshufflings ] arg Number of reshufflings, default 100
-s [ --seed ] arg Random number generator seed, default 42
--progress Display ASCII progress bar on stderr
Most options are the same as in the "classic" MULTOVL. The free regions file must be specified using the -F option, it can have any of the supported input file formats. Fixed tracks may be defined by the -f option which can be repeated several times, one for each fixed file. The number of reshufflings is controlled by the -r option. It is advisable to start with a small number and then go up to at least 1000 reshufflings for a production run. If you specify the --progress option then a simple ASCII progress bar is printed to the standard error, which is useful if you have requested lots of reshufflings. The random number generator seed can be defined by the -s option. The multovlprob runs are deterministic in the sense that the program always starts from a well-defined random seed.
The output of multovlprob contains only the statistical information. If you need the actual overlap results with the unshuffled input tracks, run the "classic" multovl beforehand. Here is a sample output:
# Parameters = -L 1 -m 2 -M 0 -F ../../src/test/data/triplefree.bed \
-r 100 -s 42 -f ../../src/test/data/triplea.bed
# Input files = 3
# ../../src/test/data/triplea.bed = track 1, region count = 2 [fixed]
# ../../src/test/data/tripleb.bed = track 2, region count = 2 [shuffled]
# ../../src/test/data/triplec.bed = track 3, region count = 2 [shuffled]
# == Overlap length statistics ==
Multiplicity,Actual,Mean,SD,Pvalue,Zscore
2,200,263.44,104.863,0.07,-0.604978
3,202,94.4062,71.9305,0.443956,1.4958
4,0,56.5,44.3286,0,-1.27457
Here the triplea.bed track was held fixed. First the settings are listed, then the overlap length statistics is printed in CSV format. The "Actual" column lists the total number of overlapping basepairs without shuffling. "Mean" and "SD" are the mean and standard deviation of the null distributions, respectively. The "Pvalue" column is a naive two-sided p-value approximation: it is the CDF("Actual") estimate if it is below 0.5, or 1.0-CDF("Actual") otherwise. Small values indicate "significant" overlaps. The "Zscore" column shows ("Actual"-"Mean")/"SD". In the example above, the pairwise total overlap length is "significant" at p < 0.1, the triple overlaps could have happened by chance. The last row shows that no fourfold overlap was detected in the unshuffled data, but some did occur during the reshufflings. The interpretation of these results is left as an exercise to the reader.
The repeated reshuffling operations in multovlprob can be speeded up near-linearly on multicore machines by assigning them to more than one thread. This is implemented in the program parmultovlprob. It takes exactly the same command-line arguments as multovlprob and launches as many worker threads at the reshuffling step as there are CPU cores. You may override this by specifying the number of worker threads using the -T threadno option. Note that each worker thread holds a private copy of the input track data structures: be aware of the speed/memory tradeoff.
Although MULTOVL can read BED-formatted files directly, it can be useful to convert them to GFF format so that the BED→GFF coordinate conversion is also applied. Here is how to use it:
This script converts one or more BED files to GFF files.
Usage: bed2gff.sh [options] file1.bed [file2.bed ...]
Options:
-c Apply BED->GFF coordinate transformation (default: no change)
-o Overwrite output file(s) if they exist (default: skip)
-h Print this help and exit
Each dir/path/file.bed will be saved to file.gff in the current directory
For instance if you issue the command:
bed2gff.sh -c -o foo/file1.bed bar/file2.bed
then the script will write file1.gff and file2.gff in the current directory, converting the BED coordinates to the GFF convention, and overwriting the output files if they exist.
Sometimes you would like to list the ancestor regions from one of the input tracks that overlapped with the others. This little script helps you do this. Invoke it as follows:-
anctrack.awk -v trackid=X ovl.gff
where X is the parent track ID, an integer 1,2, ... and multovl.gff is the output of a MULTOVL run. The ancestor regions from the specified parent track are printed to the standard output in BED format. Note that it is possible that an ancestor region participated in more than one overlap. It is advisable to run the result through the UNIX tools sort and uniq to obtain a unique list of regions like this:-
anctrack.awk -v trackid=X ovl.gff | sort | uniq > Xancestor.gff
The following tools are provided to test/exercise MULTOVL:
The utility inputfiles can generate synthetic tracks with groups of overlapping regions. Usage:
Input files for multovl testing
Usage: inputfiles [options]
Options:
-c use N chromosomes (default 2)
-d regions within overlap groups are shifted by N (default 10)
-D output directory (mandatory)
-t generate N tracks (default 3)
-g make N overlap groups (default 4)
-l region length (default 30)
-L gap between groups (default 100)
-h|-?: print this help and exit
Writes the files track_{0,1,...}.bed and free.bed to output directory
Example: regions generated for -c 1 -g 2 -t 3
reglen
<---------->
: :
Track 1 |==========| |==========|
Track 2 : |==========| : |==========|
Track 3 : : |==========| : |==========|
: : : :
<---> <------------>
delta groupgap
The script runner.sh exercises a MULTOVL tool by running it repeatedly on a set of input files made by inputfiles. This can be used for timing measurements. Usage:
Script to run a MULTOVL variant with big input files 'forever'
Usage: runner.sh [script-options] program [program-options]
Script options:
-c : number of chromosomes in input files, default 20
-d : relative shift of regions, default 10
-g : number of overlap groups, default 1000
-t : number of input tracks, default 8
-h, -?: Print this help and exit
The MULTOVL program accepts text files in BED or GFF format. Only the chromosome, first/last position, region name (if present) and strand information is parsed. The program can also read BAM files using the BAMtools library. When reading from BAM files even the region names are ignored in the interest of speed.
Be aware that MULTOVL always uses the "GFF coordinate system" convention. In other words, regions are closed intervals where the start and end coordinates correspond to the nucleotides, as opposed to the "BED coordinate system" convention where the start and end positions would correspond to the inter-nucleotide position before the first and after the last nucleotide within the region, respectively. Even if you supply BED files as input, MULTOVL interprets the start and end positions there according to the GFF convention. Use the bed2gff.sh script to convert BED coordinates to GFF.
Note that MULTOVL uses the extremely simple-minded "strategy" of deducing the file format from the input file extension. So please call your BED files something.bed, your GFF files something.gff, and your BAM files something.bam -- you get the idea.
Also note that if one of the input files has a format MULTOVL cannot detect, you get an error message and the program stops. Please make sure all input files are parsable by MULTOVL.
The MULTOVL output file format is GFF2 by default, with some additional goodies. Here is an example. Note that the line numbers are not part of the actual output, I added them by hand for the sake of the explanations below:-
1 ##gff-version 2
2 ##date 2016-04-15
3 ##source-version Multovl version 1.3, Release build, compiler: Clang version 7.0.2.7000181, system: Darwin 14.5.0 x86_64 (64-bit)
4 # Parameters = -L 1 -m 2 -M 0 -s multovl -f GFF
5 # Input files = 2
6 # ../../../src/test/data/rega.bed = track 1, region count = 8
7 # ../../../src/test/data/regb.bed = track 2, region count = 9
8 # Overlap count = 8
9 # Multiplicity counts = 1,2:7 2:1
10 chr1 multovl overlap 100 150 2 . . ANCESTORS 1:REGa:.:100-200|2:REGb:.:50-150
11 chr1 multovl overlap 250 300 2 . . ANCESTORS 1:REGa:.:250-300|2:REGb:.:250-300
12 chr1 multovl overlap 340 340 2 . . ANCESTORS 1:REGa:.:340-340|2:REGb:.:340-340
13 chr1 multovl overlap 400 410 2 . . ANCESTORS 1:REGa:.:350-450|2:REGb:.:400-410
14 chr1 multovl overlap 415 415 2 . . ANCESTORS 1:REGa:.:350-450|2:REGb:.:415-415
15 chr1 multovl overlap 420 430 2 . . ANCESTORS 1:REGa:.:350-450|2:REGb:.:420-430
16 chr1 multovl overlap 440 450 2 . . ANCESTORS 1:REGa:.:350-450|2:REGb:.:440-500
17 chr1 multovl overlap 490 500 2 . . ANCESTORS 2:REGb:.:440-500|2:REGb:.:490-510
Let's walk through this line-by-line:
The multiplicity of the overlap is indicated in Column 6 ("score"). If you searched for union overlaps, then this field shows the maximal multiplicity seen in the union, and this number can be less than the total number of ancestor regions participating in the overlap.
The ancestry information is listed in Column 9 as the attribute "ANCESTORS". The value consists of ancestor description strings, which are concatenated using the pipe symbol |, have the format [rep*]trackid:name:strand:first-last. For instance the string 2:REGb:.:50-150 represents an ancestor region in Track 2 with the name 'RegB', unknown strand orientation, from position 50 to 150. (The chromosome needs not be specified as it is the same as that of the descendant region.) The optional prefix rep* where rep is an integer > 1 indicates ancestor regions that occurred more than once in the input file. For instance 3*2:REGb:.:50-150 shows that there were 3 REGb regions in track 2 with exactly the same coordinates. The ancestors are listed in increasing track ID order, within the same track regions with lower start positions come first.
The BED output format is very similar. It begins with exactly the same comments as the GFF output (lines 4 to 9 above), and then the regions are listed. The ancestry information is provided in Column 4 (the "name" field), the multiplicity in Column 5 ("score").
I would like to thank my colleagues Markus Jaritz, Johanna Trupke, Roman Stocsits for providing me with use cases, feature requests and bug reports while developing MULTOVL. Special thanks are due to Meinrad Busslinger for his support. This work was partially financed by the Gen-AU epigenetics consortium.